Neoplasia

On this page
🧬 The Cancer Genesis Blueprint - Foundations of Neoplastic Transformation
Neoplasia represents one of medicine's most complex challenges: normal cells hijacking their own regulatory machinery to become immortal, invasive, and lethal. Understanding neoplasia means mastering the molecular switches, cellular behaviors, and systemic consequences that transform tissue architecture into chaos. This lesson builds your framework from nomenclature through molecular mechanisms to clinical detection-equipping you with the logic to predict tumor behavior, interpret markers, and recognize cancer's systemic fingerprints. Master these principles, and you'll decode every oncologic presentation with precision.

Neoplasia means "new growth"-an autonomous proliferation of cells that persists even after the initiating stimulus is removed. Unlike hyperplasia (which reverses when stimulus ends), neoplastic growth is irreversible and monoclonal (arising from a single transformed cell). Every cancer begins with genetic damage accumulating over 5-20 years, progressing through initiation, promotion, and progression phases.
⭐ Clinical Pearl: The average solid tumor contains 10⁹ cells (1 gram mass, 1 cm³ volume) at the time of clinical detection-representing approximately 30 population doublings from the original transformed cell. Most cancers are clinically silent for 75% of their natural history.
Tumor Architecture: Components and Characteristics
Every neoplasm contains two essential components:
- Parenchyma - the transformed neoplastic cells
- Determines tumor behavior and classification
- Provides the tumor's name (e.g., adenocarcinoma = malignant glandular cells)
- Clonal population with accumulated mutations
- Stroma - the supporting framework
- Connective tissue, blood vessels, immune cells
- Derived from host, not neoplastic
- Provides nutrients via angiogenesis
- Can constitute >50% of tumor mass (desmoplastic response)
- Critical for metastatic cascade support
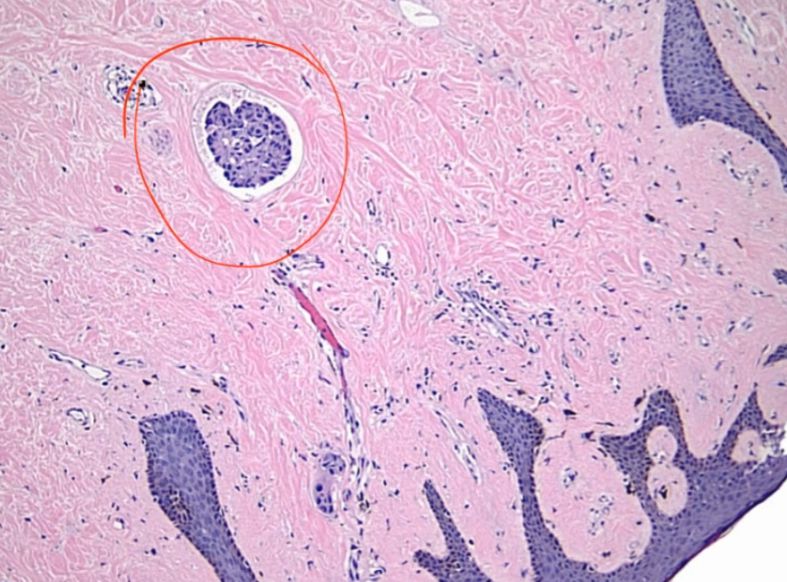
💡 Master This: Desmoplasia (dense fibrous stroma) creates the "hard" consistency of scirrhous carcinomas like breast cancer. The stroma-to-parenchyma ratio determines tumor consistency-high stromal content produces firm, gritty masses; low stromal content creates soft, fleshy tumors. This tactile difference guides surgical planning and biopsy approaches.
Neoplastic Growth Patterns: Clonality and Heterogeneity
- Monoclonality - all tumor cells derive from one transformed progenitor
- Proven by X-chromosome inactivation studies (HUMARA assay)
- Glucose-6-phosphate dehydrogenase (G6PD) isoenzyme patterns
- Immunoglobulin gene rearrangements in lymphomas
- >95% of cancers are monoclonal at origin
- Tumor heterogeneity develops during progression
- Genetic instability generates subclones with different properties
- Explains variable drug resistance within single tumors
- Creates 10⁶-10⁷ mutations per tumor genome
- Drives metastatic potential variation among cells
📌 Remember: CLONE for tumor origin characteristics - Clonal expansion, Loss of growth control, Oncogene activation, Neovascularization required, Evades apoptosis. Every cancer starts as one cell but evolves into a genetically diverse population through ongoing mutation.
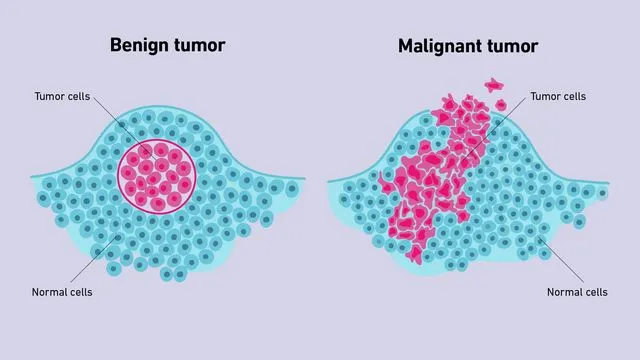
| Feature | Benign | Malignant | Clinical Significance |
|---|---|---|---|
| Growth rate | Slow, progressive | Rapid, erratic | Doubling time: 60-90 days (benign) vs 30-60 days (malignant) |
| Differentiation | Well-differentiated | Variable anaplasia | Grade determines prognosis; well-differentiated = 70-90% 5-year survival |
| Mitoses | Rare, normal | Frequent, atypical | >10 mitoses/HPF suggests malignancy in most tissues |
| Nuclear features | Normal N:C ratio | ↑ N:C ratio, hyperchromasia | N:C ratio >1:4 (vs normal 1:6) indicates malignancy |
| Capsule | Present, well-defined | Absent or infiltrative | Encapsulation enables complete surgical excision |
| Metastasis | Never | Common (30-50% at diagnosis) | Single most reliable criterion of malignancy |
The distinction between benign and malignant neoplasms determines treatment strategy, prognosis, and survival. Understanding these foundational principles prepares you to master tumor nomenclature and classification systems.
🧬 The Cancer Genesis Blueprint — Foundations of Neoplastic Transformation

📛 The Naming Game - Tumor Nomenclature Mastery
Tumor nomenclature follows systematic rules that instantly communicate tissue origin, differentiation, and malignant potential. Master the naming conventions, and you'll predict tumor behavior from the diagnosis alone-translating pathology reports into clinical action plans with precision. Every tumor name encodes critical information about cell type, aggressiveness, and expected clinical course.

Benign Tumor Nomenclature: The "-oma" Suffix System
Benign tumors add -oma to the cell type of origin:
- Epithelial benign tumors
- Adenoma - glandular epithelium (colonic polyps, pituitary adenoma)
- Papilloma - epithelium with finger-like projections (skin tags, bladder papilloma)
- Cystadenoma - adenoma forming cystic spaces (ovarian cystadenoma)
- Mesenchymal benign tumors
- Fibroma - fibrous connective tissue
- Lipoma - adipose tissue (most common soft tissue tumor, 16% of all benign tumors)
- Chondroma - cartilage
- Osteoma - bone
- Leiomyoma - smooth muscle (uterine fibroids affect 70-80% of women by age 50)
- Rhabdomyoma - skeletal muscle (rare, <1% of benign tumors)
📌 Remember: FLOC for common benign mesenchymal tumors - Fibroma (connective tissue), Lipoma (fat), Osteoma (bone), Chondroma (cartilage). These four account for >60% of benign mesenchymal neoplasms encountered clinically.
Malignant Tumor Nomenclature: Carcinoma vs Sarcoma
The fundamental division separates epithelial from mesenchymal malignancies:
- Carcinoma - malignant epithelial tumor
- Adenocarcinoma - malignant glandular epithelium (colon, prostate, breast)
- Squamous cell carcinoma - malignant squamous epithelium (lung, skin, cervix)
- Transitional cell carcinoma - malignant urothelium (bladder, renal pelvis)
- Carcinomas account for 80-90% of all human cancers
- Spread primarily via lymphatics initially
- Sarcoma - malignant mesenchymal tumor
- Fibrosarcoma - malignant fibrous tissue
- Liposarcoma - malignant adipose tissue
- Leiomyosarcoma - malignant smooth muscle
- Rhabdomyosarcoma - malignant skeletal muscle
- Osteosarcoma - malignant bone-forming cells
- Chondrosarcoma - malignant cartilage
- Sarcomas represent <1% of adult cancers but 15% of pediatric malignancies
- Spread primarily via hematogenous routes
⭐ Clinical Pearl: The carcinoma-to-sarcoma ratio is approximately 90:1 in adults but only 6:1 in children. This reflects the greater proportion of mesenchymal tissue in growing children and the epithelial predominance in adult tissue mass. Sarcomas carry worse prognosis with 5-year survival of 50-60% vs 65-70% for carcinomas at comparable stages.
Special Nomenclature Categories: The Exceptions
- Teratoma - tumor from >1 germ layer (ectoderm, mesoderm, endoderm)
- Mature teratoma: well-differentiated tissues (benign, 95% cure rate)
- Immature teratoma: embryonal elements (malignant potential 20-40%)
- Most common in gonads (ovary 80%, testis 20%)
- Hamartoma - disorganized mass of native tissue in normal location
- Not true neoplasm (developmental malformation)
- Pulmonary hamartoma (most common benign lung tumor)
- No malignant potential (<1% transformation risk)
- Choristoma - normal tissue in abnormal location
- Ectopic rest (e.g., pancreatic tissue in stomach wall)
- Developmental anomaly, not neoplastic
- Blastoma - tumor resembling embryonic tissue
- Nephroblastoma (Wilms tumor) - 500 cases/year in U.S.
- Retinoblastoma - 1 in 15,000 live births
- Neuroblastoma - most common extracranial solid tumor in children
💡 Master This: The suffix "-blastoma" signals embryonal origin and pediatric age group-these tumors arise from primitive cells that failed to mature. Despite aggressive histology, many respond well to chemotherapy due to high proliferative rates, achieving cure rates of 70-90% with modern protocols.
Hematopoietic and Lymphoid Malignancies
Blood and lymphoid cancers follow different naming conventions:
- Leukemia - malignant proliferation of blood-forming cells
- Acute leukemias: >20% blasts in bone marrow
- Chronic leukemias: mature-appearing cells predominate
- ALL, AML, CLL, CML represent 90% of leukemias
- Lymphoma - solid tumor of lymphoid tissue
- Hodgkin lymphoma: 15% of lymphomas, Reed-Sternberg cells
- Non-Hodgkin lymphoma: 85% of lymphomas, diverse subtypes
- 60,000 new cases/year in U.S.
- Multiple myeloma - plasma cell malignancy
- 30,000 new cases/year in U.S.
- Median age 70 years
| Tumor Type | Benign Name | Malignant Name | Peak Age | Frequency |
|---|---|---|---|---|
| Glandular epithelium | Adenoma | Adenocarcinoma | 50-70 years | 30% of all cancers |
| Squamous epithelium | Papilloma | Squamous cell carcinoma | 60-80 years | 20% of all cancers |
| Smooth muscle | Leiomyoma | Leiomyosarcoma | 40-60 years | 5-10% of sarcomas |
| Bone | Osteoma | Osteosarcoma | 10-25 years | 20% of bone cancers |
| Adipose tissue | Lipoma | Liposarcoma | 50-65 years | 15% of sarcomas |
| Blood vessels | Hemangioma | Angiosarcoma | 60-70 years | 2% of sarcomas |
📌 Remember: ALTOS for high-yield malignant suffixes - Adenocarcinoma (glands), Lymphoma (lymphoid), Teratoma-immature (germ cells), Osteosarcoma (bone), Squamous cell carcinoma (squamous epithelium). These five categories encompass >70% of human malignancies.
Understanding nomenclature enables instant recognition of tumor origin and behavior. This foundation supports the critical skill of distinguishing benign from malignant features through gross and microscopic examination.
📛 The Naming Game — Tumor Nomenclature Mastery

🔬 Cellular Signatures - Microscopic Features of Neoplasia
Microscopic examination reveals the cellular chaos that defines malignancy: nuclear atypia, architectural disarray, and mitotic aberrations that distinguish aggressive cancers from indolent growths. Master these histologic features, and you'll grade tumors, predict behavior, and guide treatment intensity with the precision of an expert pathologist. Every microscopic detail encodes prognostic information.

Differentiation and Anaplasia: The Spectrum of Cellular Maturity
Differentiation describes how closely neoplastic cells resemble their normal counterparts:
- Well-differentiated tumors
- Cells closely mimic normal tissue architecture
- Retain specialized functions (hormone production, mucin secretion)
- Slow growth, better prognosis
- 5-year survival: 70-90% for most organ systems
- Poorly differentiated tumors
- Minimal resemblance to tissue of origin
- Loss of specialized functions
- Rapid growth, worse prognosis
- 5-year survival: 20-40% for most organ systems
- Undifferentiated (anaplastic) tumors
- No recognizable differentiation
- Tissue of origin identified only by immunohistochemistry or molecular markers
- Most aggressive behavior
- 5-year survival: <20%
⭐ Clinical Pearl: Differentiation grade inversely correlates with proliferation rate-well-differentiated tumors have doubling times of 60-90 days, while poorly differentiated cancers double every 20-40 days. This explains why high-grade tumors respond better to chemotherapy (targeting rapidly dividing cells) but relapse more quickly.
Anaplasia: The Hallmarks of High-Grade Malignancy
Anaplasia (Greek: "to form backward") represents loss of structural and functional differentiation:
- Nuclear abnormalities - most reliable indicators
- Nuclear pleomorphism: variation in nuclear size and shape
- Hyperchromasia: dark-staining nuclei (increased DNA content)
- Increased nuclear-to-cytoplasmic (N:C) ratio: >1:4 (normal 1:6-1:8)
- Coarse chromatin clumping: irregular DNA distribution
- Prominent nucleoli: often multiple, >3 μm diameter
- Cellular abnormalities
- Cellular pleomorphism: marked variation in cell size and shape
- Loss of polarity: disorganized cellular arrangement
- Tumor giant cells: cells with >20 times normal diameter
- Multinucleation: cells with 2-50+ nuclei
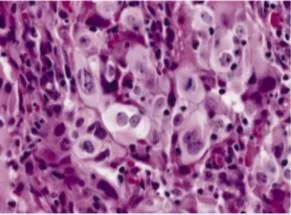
💡 Master This: The N:C ratio provides the single most reliable microscopic criterion for malignancy. Normal cells maintain N:C ratios of 1:6 to 1:8, while malignant cells show ratios approaching 1:1 in high-grade tumors. This reflects the nuclear enlargement from DNA content abnormalities and the cytoplasmic reduction from impaired differentiation.
Mitotic Activity: Quantifying Proliferation
Mitotic figures indicate proliferative activity and grade:
- Normal mitoses in benign tumors
- <2 mitoses per 10 high-power fields (HPF)
- Proper mitotic spindle formation
- Bipolar spindles, symmetric division
- Increased mitoses in malignant tumors
- >10 mitoses/10 HPF suggests high-grade malignancy
- Threshold varies by tissue (breast: >7/10 HPF = grade 3)
- Atypical mitoses - pathognomonic for malignancy
- Tripolar or multipolar spindles
- Asymmetric division
- Lagging chromosomes
- Present in 60-80% of high-grade cancers
Architectural Patterns: Loss of Normal Organization
Malignant tumors disrupt normal tissue architecture:
- Loss of normal layering
- Epithelial stratification disrupted
- Basement membrane invasion
- >50% architectural distortion in high-grade lesions
- Disorganized growth patterns
- Sheets, nests, cords replacing normal structures
- Gland formation absent or rudimentary
- Back-to-back glands without intervening stroma
- Stromal invasion
- Irregular infiltrative borders
- Desmoplastic stromal response
- Perineural invasion (seen in 30-40% of aggressive carcinomas)
- Lymphovascular invasion (strong predictor of metastasis, HR 2.5-3.0)
📌 Remember: PLAN for architectural features of malignancy - Pleomorphism (nuclear variation), Loss of polarity, Atypical mitoses, Necrosis. These four features form the foundation of histologic grading systems across all organ systems.
Tumor Necrosis: The Ischemic Signature
Necrosis indicates rapid growth outstripping blood supply:
- Coagulative necrosis - most common pattern
- Central tumor regions become ischemic
- Occurs when tumor diameter >1-2 mm (diffusion limit)
- Triggers angiogenesis via HIF-1α upregulation
- Prognostic significance
- Extensive necrosis (>50% tumor volume) indicates high grade
- Associated with 2-3 fold increased metastatic risk
- Predicts poor response to radiation (hypoxic cells resist radiation)
| Histologic Feature | Low-Grade | High-Grade | Prognostic Impact |
|---|---|---|---|
| Differentiation | Well-differentiated | Poorly differentiated/anaplastic | Grade 1 vs 3: 3-fold survival difference |
| Mitoses/10 HPF | <5 | >15 | Each 5-mitosis increase: 20% ↑ mortality |
| N:C ratio | 1:6 to 1:4 | >1:2 | High N:C: 2-fold ↑ recurrence risk |
| Necrosis | Absent | Present (>10% area) | Necrosis present: HR 2.5 for death |
| Nuclear grade | Small, uniform | Large, pleomorphic | High nuclear grade: 40% ↓ 5-year survival |
| Atypical mitoses | Absent | Present | Atypical mitoses: specific for malignancy |
⭐ Clinical Pearl: The presence of lymphovascular invasion (tumor cells within endothelial-lined spaces) predicts occult metastasis with 70-80% accuracy and independently increases mortality risk by 2.5-3.0 fold across all solid tumor types. This single finding often determines adjuvant therapy recommendations.
Grading Systems: Translating Histology to Prognosis
Histologic grading combines differentiation, mitoses, and architecture:
- Nottingham grading (breast cancer)
- Tubule formation (1-3 points)
- Nuclear pleomorphism (1-3 points)
- Mitotic count (1-3 points)
- Total 3-5 = Grade 1; 6-7 = Grade 2; 8-9 = Grade 3
- 10-year survival: Grade 1 85%, Grade 2 60%, Grade 3 40%
- Fuhrman grading (renal cell carcinoma)
- Based on nuclear size, shape, and nucleolar prominence
- Grade 1-4 scale
- 5-year survival: Grade 1 90%, Grade 4 20%
- Gleason scoring (prostate cancer)
- Architectural patterns rated 1-5
- Sum of two most common patterns (score 2-10)
- Gleason ≥8: 50% metastasis risk at 10 years
Microscopic features provide the foundation for tumor staging and clinical decision-making. Understanding cellular signatures enables recognition of aggressive biology requiring intensive treatment approaches.
🔬 Cellular Signatures — Microscopic Features of Neoplasia
🧪 The Molecular Revolution - Genetic Basis of Neoplastic Transformation
Cancer emerges from accumulated genetic damage disrupting the fundamental cellular programs governing proliferation, differentiation, and death. Master the molecular switches-oncogenes, tumor suppressors, and DNA repair systems-and you'll predict tumor behavior, interpret targeted therapy mechanisms, and understand why some cancers respond brilliantly while others resist treatment. Every cancer is fundamentally a genetic disease of somatic cells.

The Multistep Carcinogenesis Model: Accumulating Mutations
Cancer develops through sequential acquisition of 4-7 critical mutations over 10-30 years:
- Initiation - first irreversible genetic damage
- Single cell acquires mutation in growth control gene
- Requires DNA damage to critical genes (oncogenes, tumor suppressors)
- Initiated cells remain dormant without promotion
- Promotion - clonal expansion of initiated cells
- Reversible proliferative stimulus (hormones, growth factors)
- Requires 10-20 years of exposure in most solid tumors
- Generates large population of mutated cells
- Progression - additional mutations confer malignant properties
- Karyotypic instability accelerates mutation rate
- Subclones emerge with metastatic capability
- Tumor heterogeneity develops (10⁶-10⁷ mutations per tumor)
💡 Master This: The latency period between carcinogen exposure and cancer diagnosis averages 15-30 years for most solid tumors, reflecting the time required to accumulate 4-7 driver mutations. This explains why smoking cessation reduces lung cancer risk slowly-initiated cells persist for decades, requiring additional mutations to progress. Risk decreases 50% only after 10 years of cessation.
Oncogenes: Growth Signal Accelerators
Proto-oncogenes are normal genes promoting cell proliferation; mutations convert them to oncogenes with constitutive activity:
- Mechanism of activation
- Point mutations (e.g., RAS mutations in 30% of cancers)
- Chromosomal translocation (e.g., BCR-ABL in CML, 95% of cases)
- Gene amplification (e.g., ERBB2/HER2 in 20-30% breast cancer)
- Requires only one allele mutation (dominant gain-of-function)
- Major oncogene families
- Growth factors: PDGF, FGF (autocrine stimulation)
- Growth factor receptors: EGFR, HER2, KIT
- Signal transducers: RAS (mutated in 30% of cancers), ABL
- Nuclear transcription factors: MYC (amplified in 15% of cancers)
- Cell cycle regulators: Cyclins, CDKs
📌 Remember: GREAT for oncogene activation mechanisms - Gene amplification (HER2), Rearrangement/translocation (BCR-ABL), Enhancer insertion (MYC), Activating point mutation (RAS), Truncation (removes regulatory domain). These five mechanisms account for >95% of oncogene activation events.
RAS Oncogenes: The Master Switch
RAS proteins function as molecular switches in growth signaling:
- Normal RAS function
- GTPase cycling between active (GTP-bound) and inactive (GDP-bound) states
- Activates downstream MAP kinase pathway
- Intrinsic GTPase activity limits signaling duration
- RAS mutations (codons 12, 13, 61)
- Impair GTPase activity, locking RAS in active GTP-bound state
- Constitutive growth signaling independent of upstream stimuli
- KRAS: 90% of pancreatic cancers, 50% of colon cancers
- NRAS: 20% of melanomas, 15% of AML
- HRAS: 10% of bladder cancers
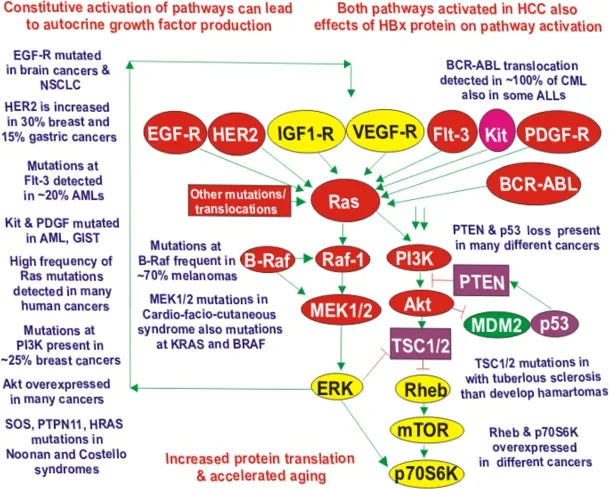
⭐ Clinical Pearl: RAS mutations predict resistance to EGFR-targeted therapy (cetuximab, panitumumab) in colorectal cancer. Patients with wild-type KRAS show 40-50% response rates to anti-EGFR antibodies, while those with KRAS mutations have <5% response rates. This mandates RAS mutation testing before prescribing EGFR inhibitors.
Tumor Suppressor Genes: The Brake System
Tumor suppressors inhibit cell proliferation; their loss of function removes growth constraints:
- Two-hit hypothesis (Knudson)
- Requires both alleles inactivated (recessive loss-of-function)
- First hit: germline mutation (inherited) or somatic mutation
- Second hit: LOH (loss of heterozygosity) via deletion, mutation, or methylation
- Explains familial cancer syndromes (germline first hit)
- Major tumor suppressor genes
- RB1 (retinoblastoma): cell cycle G1/S checkpoint, 30% of all cancers
- TP53 ("guardian of genome"): DNA damage response, 50% of all cancers
- APC (adenomatous polyposis coli): Wnt signaling, 80% of colon cancers
- BRCA1/BRCA2: DNA repair, 70% lifetime breast cancer risk if mutated
- NF1 (neurofibromin): RAS inhibitor, neurofibromatosis type 1
- VHL (von Hippel-Lindau): HIF degradation, renal cell carcinoma
TP53: The Guardian of the Genome
TP53 represents the most frequently mutated gene in human cancer:
- Normal TP53 functions
- DNA damage sensor activating cell cycle arrest (p21 induction)
- Triggers apoptosis if damage irreparable (BAX, PUMA activation)
- Prevents replication of damaged DNA
- "Molecular policeman" maintaining genomic integrity
- TP53 mutations
- Missense mutations (75%): produce dominant-negative proteins
- Loss of function: cells with DNA damage continue proliferating
- Mutated in 50% of all human cancers
- Li-Fraumeni syndrome: germline TP53 mutation, 90% cancer risk by age 60
- Clinical implications
- TP53 mutations confer chemoresistance (apoptosis pathway disrupted)
- Associated with high-grade tumors and poor prognosis
- Median survival: TP53 wild-type 48 months vs mutant 24 months in many cancers
💡 Master This: TP53 mutation status predicts chemotherapy response-tumors with wild-type TP53 retain apoptotic machinery and respond to DNA-damaging agents with 60-70% response rates, while TP53-mutant tumors show 20-30% response rates due to apoptosis resistance. This explains why some aggressive-appearing cancers respond poorly despite high proliferation rates.
DNA Repair Genes: The Maintenance Crew
Defective DNA repair accelerates mutation accumulation:
- Mismatch repair (MMR) genes
- Correct base-pairing errors during replication
- MLH1, MSH2, MSH6, PMS2 mutations cause Lynch syndrome
- 70-80% lifetime colorectal cancer risk
- Microsatellite instability (MSI-H): hallmark of MMR deficiency
- Nucleotide excision repair (NER)
- Removes bulky DNA adducts (UV-induced pyrimidine dimers)
- XP genes (xeroderma pigmentosum): 1000-fold ↑ skin cancer risk
- Homologous recombination repair
- BRCA1/BRCA2: repair double-strand breaks
- Mutations confer 50-70% breast cancer risk, 40% ovarian cancer risk
- PARP inhibitor sensitivity: synthetic lethality in BRCA-mutant tumors
| Gene | Function | Mutation Frequency | Associated Cancers | Clinical Significance |
|---|---|---|---|---|
| TP53 | DNA damage response, apoptosis | 50% all cancers | All types, especially lung, colon, breast | Predicts chemoresistance, poor prognosis |
| RB1 | G1/S checkpoint control | 30% all cancers | Retinoblastoma, osteosarcoma, SCLC | Loss enables E2F-driven proliferation |
| APC | Wnt pathway inhibitor | 80% colon cancers | Colorectal (FAP syndrome) | Earliest mutation in adenoma-carcinoma sequence |
| BRCA1/2 | Homologous recombination repair | 5-10% breast/ovarian | Breast, ovarian, pancreatic | Predicts PARP inhibitor sensitivity |
| RAS | Growth signal transduction | 30% all cancers | Pancreas, colon, lung, melanoma | Predicts anti-EGFR therapy resistance |
| MYC | Transcription factor | 15% all cancers | Burkitt lymphoma, breast, lung | Amplification indicates aggressive biology |
📌 Remember: DRAT for DNA repair gene deficiency syndromes - Defective MMR (Lynch syndrome, 80% CRC risk), Recombination repair loss (BRCA, 70% breast cancer risk), Ataxia-telangiectasia (ATM, lymphoma risk), Thymine dimer repair failure (xeroderma pigmentosum, 1000x skin cancer risk). These hereditary syndromes demonstrate how repair defects drive carcinogenesis.
⭐ Clinical Pearl: Microsatellite instability-high (MSI-H) tumors (MMR-deficient) show exceptional response to immune checkpoint inhibitors (pembrolizumab, nivolumab) with 40-60% response rates vs <10% in microsatellite-stable tumors. The high mutation burden generates neoantigens that prime anti-tumor immunity, making MSI status a critical biomarker for immunotherapy selection.
Understanding molecular mechanisms transforms cancer from a morphologic diagnosis into a targetable genetic disease. This knowledge foundation enables interpretation of tumor markers and their clinical applications.
🧪 The Molecular Revolution — Genetic Basis of Neoplastic Transformation


🎯 Cancer's Calling Cards - Tumor Markers in Clinical Practice
Tumor markers are measurable substances-proteins, hormones, or genetic material-produced by cancer cells or host responses that enable detection, monitoring, and prognostication. Master the markers, and you'll screen high-risk populations, track treatment response, and detect recurrence months before clinical or radiologic evidence emerges. Every marker has specific indications, limitations, and thresholds that guide interpretation.

Tumor Marker Principles: Sensitivity, Specificity, and Clinical Utility
No tumor marker achieves 100% sensitivity and specificity-understanding their limitations prevents misuse:
- Sensitivity - ability to detect cancer when present
- High sensitivity needed for screening (minimize false negatives)
- Most markers have 60-80% sensitivity (miss 20-40% of cancers)
- Specificity - ability to exclude cancer when absent
- High specificity needed to avoid false positives (unnecessary workup)
- Elevated in benign conditions (10-30% false positive rate)
- Positive predictive value (PPV) - probability of cancer given positive test
- Depends on disease prevalence
- Low in screening general population (PPV <10% for most markers)
💡 Master This: Tumor markers are not suitable for screening average-risk populations due to low PPV-even with 95% specificity, screening 1000 people with 1% cancer prevalence yields 10 true positives but 50 false positives, requiring extensive workup for benign conditions. Markers excel in monitoring known cancer where high pretest probability improves PPV to >80%.
Serum Tumor Markers: The Major Players
- Alpha-fetoprotein (AFP)
- Normal: <10 ng/mL
- Elevated in: hepatocellular carcinoma (60-70% of cases), nonseminomatous germ cell tumors (yolk sac tumor)
- Threshold: >400 ng/mL strongly suggests HCC
- Monitoring: rising AFP predicts recurrence 3-6 months before imaging
- False positives: cirrhosis, hepatitis, pregnancy
- Carcinoembryonic antigen (CEA)
- Normal: <3 ng/mL (nonsmokers), <5 ng/mL (smokers)
- Elevated in: colorectal cancer (70% of cases), pancreatic, gastric, lung
- Threshold: >10 ng/mL suggests malignancy
- Monitoring: detects 70% of recurrences before symptoms
- False positives: smoking, inflammatory bowel disease, pancreatitis
- CA 19-9 (carbohydrate antigen 19-9)
- Normal: <37 U/mL
- Elevated in: pancreatic cancer (80% of cases), cholangiocarcinoma, gastric
- Threshold: >1000 U/mL indicates unresectable disease
- Limitation: 10% of population lacks Lewis antigen (cannot produce CA 19-9)
- False positives: biliary obstruction, pancreatitis

📌 Remember: CHAP for gastrointestinal tumor markers - CEA (colorectal, 70% sensitivity), HCC-AFP (hepatocellular, 60-70%), AFP-germ cell (yolk sac tumor, 90%), Pancreas-CA 19-9 (pancreatic, 80%). These four markers cover the major GI malignancies encountered in clinical practice.
Hormone Markers: Functional Tumor Products
Tumors producing hormones generate measurable markers:
- Human chorionic gonadotropin (β-hCG)
- Normal: <5 mIU/mL (non-pregnant)
- Elevated in: choriocarcinoma, hydatidiform mole, nonseminomatous germ cell tumors
- Threshold: >100,000 mIU/mL in choriocarcinoma
- Monitoring: half-life 24-36 hours; should normalize post-treatment
- False positives: pregnancy, pituitary hCG production
- Calcitonin
- Normal: <10 pg/mL
- Elevated in: medullary thyroid carcinoma (100% of cases)
- Threshold: >100 pg/mL diagnostic
- Screening: used in MEN 2 syndrome families
- Monitoring: undetectable calcitonin indicates cure
- Catecholamines/metanephrines
- Elevated in: pheochromocytoma (95% sensitivity)
- 24-hour urine: metanephrines >2x upper limit diagnostic
- Plasma metanephrines: 99% sensitivity, preferred screening test
Prostate-Specific Antigen (PSA): Controversy and Context
PSA represents the most widely used yet controversial tumor marker:
- Normal values (age-adjusted)
- 40-49 years: <2.5 ng/mL
- 50-59 years: <3.5 ng/mL
- 60-69 years: <4.5 ng/mL
- 70-79 years: <6.5 ng/mL
- Interpretation thresholds
- <4 ng/mL: 15% cancer risk
- 4-10 ng/mL (gray zone): 25% cancer risk
- >10 ng/mL: >50% cancer risk
- PSA velocity: >0.75 ng/mL/year suggests cancer
- PSA density: PSA/prostate volume; >0.15 ng/
Continue reading on OnCourse
Sign up for free to access the full lesson, plus unlimited questions, flashcards, AI-powered notes, and more.
CONTINUE READING — FREEor get the app
Have doubts about this lesson?
Ask Rezzy, our AI tutor, to explain anything you didn't understand
Practice Questions: Neoplasia
Test your understanding with these related questions
What is the most common carcinoma to develop after burns?