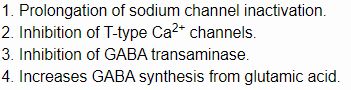Central Nervous System Pharmacology

On this page
🧠 The CNS Pharmacology Blueprint: Command & Control
Central nervous system drugs touch every major neurological disorder, from seizures to pain, sleep to addiction. Mastering CNS pharmacology means understanding how molecules cross the blood-brain barrier, modulate neurotransmitter systems, and produce therapeutic effects while navigating narrow safety windows. This lesson builds your expertise systematically-from stimulants that amplify neural signaling to anesthetics that silence consciousness, from addiction's molecular chains to antiepileptics that stabilize electrical storms.
⚡ The Psychostimulant Arsenal: Neurochemical Accelerators
Psychostimulants amplify catecholaminergic neurotransmission, driving alertness, focus, and wakefulness through dopamine and norepinephrine pathways. Understanding their mechanisms unlocks both therapeutic applications in ADHD and narcolepsy, and the addiction potential that makes them controlled substances.
Amphetamine Class Mechanisms
Amphetamines don't merely block reuptake-they reverse monoamine transporters, forcing presynaptic neurons to dump dopamine, norepinephrine, and serotonin into synaptic clefts. This produces 3-4 fold increases in extracellular catecholamines.
- Amphetamine & Dextroamphetamine
- Release mechanism: Competitive inhibition of VMAT2 (vesicular monoamine transporter-2)
- Dopamine release ↑ 300-400% in nucleus accumbens
- Duration: 4-6 hours for immediate-release formulations
- Therapeutic index: Narrow, with toxicity emerging at 2-3× therapeutic doses
- Methamphetamine
- CNS penetration: 10× greater lipophilicity than amphetamine
- Neurotoxicity threshold: Chronic use at >40 mg/day
- Dopaminergic terminal damage documented after 6-12 months heavy use

📌 Remember: AMPHETAMINE mechanism-Augments Monoamine release, Prevents reuptake, Hyperactivates Efflux via Transporter Alteration, Makes Increased Neurotransmitter Exposure
Methylphenidate Pharmacology
Methylphenidate selectively blocks dopamine and norepinephrine reuptake without triggering neurotransmitter release, producing cleaner stimulation with lower abuse potential than amphetamines.
- Mechanism Distinctions
- DAT (dopamine transporter) affinity: Ki = 34 nM
- NET (norepinephrine transporter) affinity: Ki = 339 nM
- Preferential dopamine effect in prefrontal cortex and striatum
- No serotonergic activity (unlike amphetamines)
- Clinical Formulations
- Immediate-release: Peak 1-2 hours, duration 3-4 hours
- Extended-release: Bimodal release, duration 8-12 hours
- Transdermal patch: Steady-state over 9 hours, reduces GI side effects by 40-50%
⭐ Clinical Pearl: Methylphenidate shows 70-80% response rates in ADHD versus 60-70% for amphetamines, but amphetamines demonstrate superior efficacy in treatment-resistant cases with effect sizes of 0.8-1.0 versus 0.6-0.7 for methylphenidate.
Modafinil & Wakefulness Promotion
Modafinil represents a unique stimulant class with unclear primary mechanism but documented effects on multiple neurotransmitter systems without amphetamine-like dopamine surges.
- Proposed Mechanisms
- Weak DAT inhibition: IC50 = 4 μM (much weaker than methylphenidate)
- Orexin/hypocretin pathway activation in lateral hypothalamus
- Histamine release ↑ in tuberomammillary nucleus
- GABA inhibition ↓ in sleep-promoting regions
- Pharmacokinetics
- Half-life: 12-15 hours (permits once-daily dosing)
- Hepatic metabolism: CYP3A4/5 (induces own metabolism)
- Steady state: Achieved after 2-4 days
- Clinical Applications
- Narcolepsy: 200-400 mg/day, reduces sleep attacks by 50-70%
- Shift work disorder: 150-200 mg before shift
- Obstructive sleep apnea adjunct: 200 mg/day with CPAP

💡 Master This: Modafinil's low abuse liability (Schedule IV versus Schedule II for amphetamines) stems from gradual dopamine elevation without euphoric peaks-extracellular dopamine rises only 50-60% versus 300-400% with amphetamines, and lacks the rapid onset that drives addiction.
| Feature | Amphetamine | Methylphenidate | Modafinil | Atomoxetine | Caffeine |
|---|---|---|---|---|---|
| Primary MOA | NE/DA releaser | DAT/NET blocker | Orexin/DA/His | Selective NET blocker | Adenosine antagonist |
| Abuse Potential | High (Schedule II) | Moderate (Schedule II) | Low (Schedule IV) | None | Minimal |
| ADHD Efficacy | Effect size 0.8-1.0 | Effect size 0.6-0.7 | Not indicated | Effect size 0.6-0.7 | Not indicated |
| Duration | 4-6 hours (IR) | 3-4 hours (IR) | 12-15 hours | 5-6 hours | 3-5 hours |
| Cardiovascular Risk | ↑↑↑ (HR +10-20 bpm) | ↑↑ (HR +5-10 bpm) | ↑ (minimal) | ↑ (HR +5-10 bpm) | ↑ (dose-dependent) |
| Weight Effect | ↓↓↓ (-3 to -5 kg) | ↓↓ (-2 to -3 kg) | ↓ (-1 to -2 kg) | ↓ (-1 to -2 kg) | None |
Stimulant Adverse Effect Profiles
Sympathomimetic toxicity dominates psychostimulant adverse effects, with cardiovascular and psychiatric complications limiting use in vulnerable populations.
- Cardiovascular Concerns
- Blood pressure elevation: 5-10 mmHg systolic, 3-7 mmHg diastolic
- Heart rate increase: 5-20 bpm depending on agent
- QTc prolongation: Risk with amphetamines > methylphenidate
- Sudden cardiac death: Estimated 0.5-2 per 100,000 patient-years (controversial)
- Contraindications: Structural heart disease, severe hypertension, hyperthyroidism
- Psychiatric Complications
- New-onset psychosis: 0.1-0.25% with therapeutic doses
- Mania precipitation: 5-10% in bipolar disorder
- Anxiety exacerbation: 10-20% of patients
- Irritability and mood lability: 20-30% in children
- Growth & Metabolic Effects
- Height suppression: 1-2 cm over 2-3 years with continuous use
- Weight loss: 2-5 kg in first 6-12 months
- Appetite suppression: 30-50% of patients
⚠️ Warning: Stimulant-induced psychosis typically emerges with chronic high-dose abuse (methamphetamine >100 mg/day) but can occur at therapeutic doses in genetically susceptible individuals-symptoms resolve within 7-10 days of discontinuation in 90% of cases.
📌 Remember: STIMULANT toxicity-Sympathomimetic crisis, Tachycardia/hypertension, Insomnia, Mania/psychosis, Underweight, Loss of appetite, Addiction risk, Neurological (seizures), Tremor
Connect the addiction neurobiology of stimulants through understanding how chronic dopamine pathway activation reshapes reward circuitry in the next section on cognitive enhancers and analeptics.
⚡ The Psychostimulant Arsenal: Neurochemical Accelerators

🔋 Cognitive Enhancers & Analeptics: Wakefulness Without Addiction
Beyond psychostimulants lie agents that enhance cognition or respiratory drive through distinct mechanisms-methylxanthines blocking adenosine, cholinesterase inhibitors boosting acetylcholine, and respiratory stimulants activating brainstem chemoreceptors. These drugs offer therapeutic benefits with different safety profiles.
Methylxanthine Pharmacology
Caffeine, theophylline, and theobromine antagonize adenosine receptors, preventing the accumulation of this endogenous sleep-promoting nucleoside. Adenosine normally inhibits neural firing and promotes drowsiness-blocking its A1 and A2A receptors produces wakefulness.
- Caffeine Mechanisms
- A1 receptor antagonism: Ki = 12 μM (blocks presynaptic inhibition)
- A2A receptor antagonism: Ki = 2.4 μM (disinhibits dopamine pathways)
- Phosphodiesterase inhibition: Weak effect at therapeutic doses
- Intracellular calcium mobilization: Contributes to cardiac/skeletal muscle effects
- Pharmacokinetics
- Oral bioavailability: >99% (complete absorption)
- Peak plasma: 30-60 minutes after ingestion
- Half-life: 3-7 hours (highly variable, CYP1A2-dependent)
- Metabolism: Hepatic demethylation to paraxanthine (80%), theobromine, theophylline
- Clinical Effects
- Alertness dose: 50-200 mg (equivalent to 1-2 cups coffee)
- Performance enhancement: 3-5 mg/kg improves endurance by 10-15%
- Tolerance: Develops to cardiovascular effects in 1-4 days, partial tolerance to CNS effects

⭐ Clinical Pearl: Caffeine withdrawal produces headache in 50% of regular users (>200 mg/day) within 12-24 hours of cessation, peaking at 20-48 hours-symptoms resolve with 50-100 mg caffeine or spontaneously over 2-9 days.
Theophylline: Bronchodilator with CNS Effects
Theophylline shares caffeine's adenosine antagonism but adds significant phosphodiesterase inhibition, producing bronchodilation and respiratory stimulation alongside CNS activation. Its narrow therapeutic index demands careful monitoring.
- Therapeutic Applications
- Asthma/COPD: Target range 10-20 μg/mL (bronchodilation)
- Apnea of prematurity: 5-10 μg/mL (stimulates respiratory drive)
- Mechanism: cAMP elevation → smooth muscle relaxation
- Toxicity Profile
- Therapeutic: 10-20 μg/mL
- Mild toxicity: 20-30 μg/mL (nausea, tremor, tachycardia)
- Severe toxicity: >30 μg/mL (seizures, arrhythmias)
- Lethal: >80-100 μg/mL
- Drug Interactions
- CYP1A2 inducers (smoking, rifampin): ↓ levels by 30-50%
- CYP1A2 inhibitors (ciprofloxacin, fluvoxamine): ↑ levels by 50-100%
- Macrolides (erythromycin): ↑ levels by 25-40%
💡 Master This: Theophylline's 10-fold variation in clearance between individuals-smokers metabolize 50-100% faster than nonsmokers, neonates 50% slower than adults, and elderly patients 30-40% slower-explains why therapeutic drug monitoring is mandatory for safe use.
Cholinesterase Inhibitors for Dementia
Donepezil, rivastigmine, and galantamine inhibit acetylcholinesterase in the CNS, elevating acetylcholine concentrations in cortical and hippocampal synapses. This partially compensates for cholinergic neuron loss in Alzheimer's disease.
- Donepezil Characteristics
- Selectivity: AChE >1000-fold over BuChE (butyrylcholinesterase)
- Half-life: 70 hours (once-daily dosing)
- Dose: 5-10 mg/day (start low, titrate after 4-6 weeks)
- Efficacy: MMSE improvement 2-3 points over placebo at 6 months
- Duration: Benefits persist 12-18 months, then decline
- Rivastigmine Profile
- Dual inhibition: AChE and BuChE (broader effect)
- Half-life: 1.5 hours (twice-daily dosing, or transdermal patch)
- Dose: 3-6 mg twice daily oral, or 9.5-13.3 mg/24h patch
- GI tolerability: 30-40% nausea with oral, 10-15% with patch
- Galantamine Mechanism
- AChE inhibition plus nicotinic receptor allosteric modulation
- Half-life: 7 hours (once or twice daily)
- Dose: 16-24 mg/day
- Cognitive benefit: Similar to donepezil (2-3 MMSE points)
📌 Remember: CHOLINESTERASE inhibitor effects-Cognition improved modestly, Heart rate slowed (bradycardia risk), Ocular (miosis), Lacrimation, Increased GI motility (diarrhea), Nausea/vomiting, Excessive salivation, Sweating, Tremor, Excitation then fatigue, Respiratory secretions, Anxiety, Sleep disturbance, Excessive urination
| Agent | AChE Selectivity | Half-Life | Dosing Frequency | MMSE Benefit | GI Tolerability | Unique Feature |
|---|---|---|---|---|---|---|
| Donepezil | High (>1000:1) | 70 hours | Once daily | +2 to +3 points | Good (15-20% nausea) | Longest action |
| Rivastigmine | Dual AChE/BuChE | 1.5 hours | Twice daily or patch | +2 to +3 points | Poor oral (30-40% nausea) | Patch option |
| Galantamine | Moderate + nAChR | 7 hours | Once or twice daily | +2 to +3 points | Moderate (20-25% nausea) | Dual mechanism |
| Tacrine | Low | 2-4 hours | Four times daily | +2 to +4 points | Very poor (40-50% nausea) | Hepatotoxic (obsolete) |
Respiratory Stimulants & Analeptics
Doxapram and older agents like nikethamide stimulate medullary respiratory centers, increasing respiratory rate and tidal volume. Modern use is extremely limited due to safer alternatives (mechanical ventilation, reversal agents).
- Doxapram Mechanism
- Peripheral chemoreceptor activation (carotid bodies)
- Central respiratory center stimulation (medulla)
- Dose-dependent effect: Low doses → peripheral, high doses → central
- Clinical Context
- Historical use: Postoperative respiratory depression, COPD exacerbations
- Current role: Virtually obsolete in developed settings
- Dose: 0.5-1.5 mg/kg IV bolus, then 1-3 mg/min infusion
- Duration: 5-12 minutes after bolus
- Adverse Effects
- Hypertension: 20-30 mmHg systolic elevation
- Tachycardia: 15-25 bpm increase
- Seizures: Risk with doses >3 mg/kg
- Arrhythmias: Ventricular ectopy in 5-10%
⚠️ Warning: Analeptics like doxapram produce generalized CNS excitation without selectivity for respiratory centers-seizure risk at 2-3× therapeutic doses makes them dangerous compared to naloxone for opioid-induced respiratory depression or flumazenil for benzodiazepine reversal.
Understanding how drugs enhance cognition sets the stage for examining the flip side-how substances hijack reward pathways to produce addiction, explored next through the neurobiology of substance abuse.
🔋 Cognitive Enhancers & Analeptics: Wakefulness Without Addiction

🔗 Addiction Neurobiology: The Reward Circuitry Hijack
All addictive substances converge on the mesolimbic dopamine pathway, producing supraphysiological dopamine surges in the nucleus accumbens that dwarf natural rewards. Understanding addiction's molecular basis explains tolerance, dependence, withdrawal, and relapse vulnerability.
The Mesolimbic Reward Pathway
Dopaminergic neurons originating in the ventral tegmental area (VTA) project to nucleus accumbens, prefrontal cortex, and amygdala-this circuit evolved to reinforce survival behaviors (food, sex, social bonding) but becomes pathologically activated by drugs.
- Normal Reward Processing
- Baseline dopamine: 4-8 nM in nucleus accumbens
- Food reward: Dopamine ↑ 50-100% above baseline
- Sexual activity: Dopamine ↑ 100-150%
- Social bonding: Dopamine ↑ 50-75%
- Duration: 15-30 minutes, then return to baseline
- Drug-Induced Dopamine Surges
- Cocaine: Dopamine ↑ 300-400% (blocks reuptake)
- Amphetamine: Dopamine ↑ 300-500% (reverses transporters)
- Heroin: Dopamine ↑ 200-300% (disinhibits VTA via μ-opioid receptors)
- Nicotine: Dopamine ↑ 150-200% (nAChR activation)
- Alcohol: Dopamine ↑ 100-200% (multiple mechanisms)
- Duration: 1-3 hours depending on drug half-life

💡 Master This: The 10-fold difference between natural rewards (+50-100% dopamine) and drugs like cocaine (+300-400%) explains why addiction overrides survival instincts-the brain's reward system interprets drug use as more important than food, safety, or reproduction.
Neuroadaptation & Tolerance
Chronic drug exposure triggers compensatory downregulation of dopamine signaling-receptor density decreases, signal transduction weakens, and baseline dopamine drops below normal. This creates tolerance (requiring higher doses) and anhedonia (inability to feel pleasure without the drug).
- Molecular Changes
- D2 receptor density: ↓ 15-30% after 6-12 months cocaine use
- Dopamine synthesis: ↓ tyrosine hydroxylase expression by 20-40%
- Transporter upregulation: DAT density ↑ 30-50% (faster dopamine clearance)
- CREB activation: Transcription factor changes persist weeks to months
- Functional Consequences
- Reward threshold elevation: Natural rewards produce 50-70% less dopamine response
- Anhedonia: 70-85% of cocaine users report during abstinence
- Tolerance timeline: Develops over days to weeks depending on drug
- Reversal: Partial recovery over months to years, never complete
⭐ Clinical Pearl: PET imaging reveals 20-30% lower D2 receptor availability in cocaine users that persists >12 months after cessation-this protracted dopamine deficit explains why relapse rates remain 40-60% even after 1 year of abstinence.
Physical Dependence & Withdrawal
Physical dependence emerges when homeostatic adaptations to chronic drug exposure leave the nervous system dysregulated upon drug removal. Withdrawal syndromes reflect rebound hyperactivity of systems the drug suppressed or hypoactivity of systems it enhanced.
- Opioid Withdrawal Timeline
- Onset: 6-12 hours after last short-acting opioid, 24-48 hours for methadone
- Peak: 48-72 hours
- Duration: 7-10 days acute, weeks to months protracted
- Symptoms: Lacrimation, rhinorrhea, mydriasis, piloerection, diarrhea, myalgias, anxiety
- Severity: Rarely life-threatening (unlike alcohol/benzodiazepine withdrawal)
- Alcohol/Sedative Withdrawal
- Onset: 6-24 hours after last drink
- Minor symptoms: Tremor, anxiety, tachycardia, hypertension, insomnia (6-36 hours)
- Seizures: 12-48 hours (occur in 5-15% of untreated withdrawal)
- Delirium tremens: 48-96 hours, mortality 5-15% untreated
- Kindling: Each withdrawal episode worsens subsequent ones
- Stimulant Withdrawal
- Onset: Hours to days (no acute medical danger)
- Crash phase: Hypersomnia, hyperphagia, depression (1-3 days)
- Withdrawal phase: Anhedonia, anergia, anxiety (1-10 weeks)
- Extinction phase: Episodic craving (months to years)
📌 Remember: WITHDRAWAL severity hierarchy-Worst are alcohol/benzos (life-threatening), Intermediate are opioids (severe but safe), Then stimulants (uncomfortable), Hallucinogens (minimal), Delta-9-THC (mild), Rarely Acute danger from cannabis, Watch for Alcohol seizures, Life-threatening delirium tremens
Craving & Relapse Mechanisms
Addiction persists long after physical dependence resolves because drug-associated cues, stress, and negative emotional states trigger powerful cravings mediated by glutamatergic projections from prefrontal cortex and amygdala to nucleus accumbens.
- Cue-Induced Craving
- Conditioned stimuli (people, places, paraphernalia) activate same brain regions as drug itself
- Amygdala activation: 200-300% increase in fMRI studies
- Prefrontal cortex dysfunction: Impaired inhibitory control persists months
- Dopamine release: Cues trigger 50-100% dopamine surge (without drug)
- Stress-Induced Relapse
- Corticotropin-releasing factor (CRF): ↑ in extended amygdala during withdrawal
- Stress exposure: Reinstates drug-seeking in 60-70% of abstinent users
- HPA axis dysregulation: Persists 6-12 months after cessation
- Relapse Statistics
- First year: 40-60% relapse rate across all substances
- Five years: 70-80% have relapsed at least once
- Long-term recovery: <30% maintain continuous abstinence >5 years without treatment

💡 Master This: Addiction is a chronic relapsing brain disease, not a moral failure-neuroimaging demonstrates persistent structural changes including 5-10% prefrontal cortex volume reduction and 15-20% decreased gray matter density in anterior cingulate that predict relapse with 70-80% accuracy.
With addiction's neural architecture mapped, shift focus to how antiepileptic drugs stabilize the opposite extreme-excessive synchronized neural firing that produces seizures.
🔗 Addiction Neurobiology: The Reward Circuitry Hijack

⚡ Antiepileptic Drug Mechanisms: Electrical Storm Controllers
Seizures arise from imbalanced excitation and inhibition-excessive glutamatergic drive or insufficient GABAergic restraint produces hypersynchronous neuronal firing. Antiepileptic drugs (AEDs) restore balance through five core mechanisms, each targeting different aspects of neuronal excitability.
Sodium Channel Blockade: The Frontline Mechanism
Voltage-gated sodium channels initiate action potentials-AEDs that prolong channel inactivation preferentially suppress high-frequency repetitive firing characteristic of seizures while sparing normal neuronal activity.
- Phenytoin Pharmacology
- Mechanism: Binds inactivated Na+ channels, prolongs refractory period
- Use-dependence: Blocks rapidly firing neurons >10-fold more than slow
- Therapeutic range: 10-20 μg/mL (free 1-2 μg/mL)
- Protein binding: 90% (albumin)
- Zero-order kinetics: Saturable metabolism at therapeutic doses
- Half-life: 12-36 hours (dose-dependent due to saturation)
- Carbamazepine Profile
- Mechanism: Similar Na+ channel blockade, also adenosine potentiation
- Indications: Focal seizures, trigeminal neuralgia, bipolar disorder
- Autoinduction: Induces own metabolism over 3-5 weeks, requiring dose ↑
- Therapeutic range: 4-12 μg/mL
- Half-life: 12-17 hours initially, decreases to 8-12 hours with chronic use
- Lamotrigine Advantages
- Mechanism: Na+ channel blockade plus Ca²⁺ channel effects
- Broad spectrum: Effective focal and generalized seizures
- Tolerability: Lower cognitive impairment than older AEDs
- Half-life: 25-30 hours (once or twice daily)
- Rash risk: 10% benign rash, 0.1% Stevens-Johnson syndrome
⭐ Clinical Pearl: Phenytoin exhibits zero-order kinetics above 10-15 μg/mL-small dose increases produce disproportionately large concentration rises. A 10% dose increase can raise levels by 50-100%, causing toxicity (ataxia, nystagmus, confusion) within days.
GABA Enhancement: Inhibitory Amplification
Enhancing GABAergic inhibition suppresses seizures by hyperpolarizing neurons and raising the threshold for action potential generation. Multiple AEDs target different points in GABA neurotransmission.
- Benzodiazepines (Acute Seizures)
- Mechanism: Positive allosteric modulation of GABAA receptors (↑ opening frequency)
- Lorazepam: 4 mg IV, duration 12-24 hours, preferred for status epilepticus
- Diazepam: 10 mg IV, duration 2-4 hours (rapid redistribution)
- Midazolam: 10 mg IM, alternative when IV access unavailable
- Tolerance: Develops over days to weeks, limits chronic use
- Barbiturates (Phenobarbital)
- Mechanism: GABAA receptor modulation (↑ opening duration) plus Na+ channel blockade
- Dose: 60-180 mg/day adults, 3-5 mg/kg/day children
- Half-life: 80-120 hours (once-daily dosing)
- Efficacy: 50-60% seizure freedom in focal epilepsy
- Limitations: Sedation, cognitive impairment, enzyme induction
- Vigabatrin (GABA Transaminase Inhibitor)
- Mechanism: Irreversible GABA-T inhibition → ↑ GABA levels 200-300%
- Indication: Infantile spasms (first-line), refractory focal epilepsy
- Efficacy: 50-70% response in infantile spasms
- Toxicity: 30-40% develop irreversible peripheral visual field defects
- Monitoring: Perimetry every 3-6 months

💡 Master This: Benzodiazepines and barbiturates both enhance GABAA receptors but differ critically-benzodiazepines increase opening frequency (have a ceiling effect, safer) while barbiturates prolong opening duration (no ceiling, can cause fatal respiratory depression at high doses).
Calcium Channel Modulation: Absence Seizure Specialists
T-type calcium channels in thalamic neurons generate rhythmic oscillations-blocking these channels suppresses the 3 Hz spike-wave discharges characteristic of absence epilepsy.
- Ethosuximide
- Mechanism: Selective T-type Ca²⁺ channel blockade in thalamus
- Indication: First-line for absence seizures (ineffective for other types)
- Efficacy: 70-80% seizure freedom in childhood absence epilepsy
- Dose: 15-40 mg/kg/day in 2 divided doses
- Half-life: 40-60 hours in adults, 30-40 hours in children
- Therapeutic range: 40-100 μg/mL
- Valproate (Multi-Mechanism)
- T-type Ca²⁺ blockade plus Na+ channel effects plus GABA enhancement
- Broad spectrum: Effective all seizure types
- Dose: 15-60 mg/kg/day
- Therapeutic range: 50-100 μg/mL
- Teratogenicity: Neural tube defects 1-2%, IQ reduction 7-10 points, AVOID in pregnancy
📌 Remember: ETHOSUXIMIDE for absence-Effective Thalamic Halt, Only for Spike-wave (3 Hz), Useless for Xtra seizure types, Inhibits T-type Mediated Intrinsic oscillations, Drug of choice for Early absence
Glutamate Antagonism & Synaptic Modulation
Reducing excitatory glutamatergic transmission provides another avenue for seizure suppression, though direct NMDA antagonists cause unacceptable psychotomimetic effects.
- Topiramate (Multiple Mechanisms)
- Na+ channel blockade
- AMPA/kainate receptor antagonism
- Carbonic anhydrase inhibition
- GABAA receptor potentiation
- Dose: 200-400 mg/day in 2 divided doses
- Efficacy: 40-50% responder rate as adjunct
- Adverse effects: Cognitive slowing (30-40%), word-finding difficulty, weight loss, kidney stones (1.5%)
- Levetiracetam (SV2A Modulation)
- Mechanism: Binds synaptic vesicle protein 2A (SV2A), modulates neurotransmitter release
- Broad spectrum: Focal and generalized seizures
- Dose: 1000-3000 mg/day in 2 divided doses
- Half-life: 6-8 hours
- Advantages: No drug interactions, renal elimination, well-tolerated
- Psychiatric effects: Irritability, depression in 5-10%
| Mechanism | Prototype Drug | Seizure Types | Key Advantage | Major Limitation | Therapeutic Range |
|---|---|---|---|---|---|
| Na+ channel block | Phenytoin | Focal, 2° generalized | Effective, IV available | Zero-order kinetics, interactions | 10-20 μg/mL |
| GABA enhancement | Valproate | All types (broad spectrum) | Most versatile AED | Teratogenic, hepatotoxic | 50-100 μg/mL |
| T-type Ca²⁺ block | Ethosuximide | Absence only | Highly effective absence | Narrow spectrum | 40-100 μg/mL |
| SV2A modulation | Levetiracetam | Focal, generalized | No interactions, well-tolerated | Psychiatric effects | No established range |
| Multiple mechanisms | Topiramate | Focal, generalized | Weight loss (migraine benefit) | Cognitive impairment | No established range |
Status Epilepticus Management Algorithm
Status epilepticus-continuous seizure activity >5 minutes or recurrent seizures without recovery-constitutes a neurological emergency with 10-20% mortality if untreated. Treatment follows a time-based protocol.
⚠️ Warning: Each 10 minutes of ongoing status epilepticus increases mortality by ~3% and permanent neurological damage risk-aggressive early treatment within 5 minutes reduces morbidity by 50-60% compared to delayed intervention.
From suppressing pathological electrical activity, transition to understanding how local anesthetics achieve the opposite goal-selectively silencing peripheral nerve conduction while preserving consciousness.
⚡ Antiepileptic Drug Mechanisms: Electrical Storm Controllers

💉 Local Anesthetic Pharmacology: Reversible Nerve Blockade
Local anesthetics (LAs) interrupt action potential propagation in peripheral nerves by blocking voltage-gated sodium channels from the intracellular side. Understanding their chemistry, pharmacokinetics, and toxicity profiles enables safe regional anesthesia.
Chemical Structure & Pharmacokinetics
All LAs share a lipophilic aromatic ring, intermediate ester or amide linkage, and hydrophilic tertiary amine. This structure determines potency, duration, and metabolism.
- Ester vs Amide Classification
- Esters (procaine, chloroprocaine, tetracaine): Metabolized by plasma pseudocholinesterase
- Amides (lidocaine, bupivacaine, ropivacaine): Metabolized by hepatic CYP450
- Allergy: Esters produce PABA metabolite (allergenic), amides rarely cause true allergy (<1%)
- Potency Determinants
- Lipid solubility: Correlates with potency (bupivacaine 30× more lipophilic than procaine)
- Protein binding: Correlates with duration (bupivacaine 95% bound, duration 3-6 hours)
- pKa: Determines onset (lower pKa → more unionized at physiologic pH → faster onset)
- Key Agent Profiles
- Lidocaine: pKa 7.9, onset 2-5 min, duration 1-2 hours, potency 1× (reference)
- Bupivacaine: pKa 8.1, onset 5-10 min, duration 3-6 hours, potency 4×
- Ropivacaine: pKa 8.1, onset 5-10 min, duration 3-5 hours, potency 3×, less cardiotoxic

📌 Remember: AMIDE local anesthetics-Lidocaine, Mepivacaine, Bupivacaine, Ropivacaine, Prilocaine, Etidocaine (note: "i" before "-caine" = amide)
Mechanism of Action & Differential Blockade
LAs must cross the nerve membrane in uncharged form, then ionize intracellularly to block Na+ channels. This pH dependence explains why they work poorly in infected (acidic) tissue.
- Use-Dependent Blockade
- LAs preferentially bind open and inactivated Na+ channel states
- Rapidly firing nerves (pain fibers) blocked before slow-firing (motor) nerves
- Explains why sensory blockade precedes motor blockade
- Fiber Sensitivity Hierarchy
- Most sensitive: Small myelinated (Aδ pain), unmyelinated (C pain/temperature)
- Intermediate: Large myelinated (Aγ proprioception, Aβ touch)
- Least sensitive: Large myelinated (Aα motor)
- Clinical sequence: Pain → temperature → touch → proprioception → motor
- Onset Modifiers
- Bicarbonate addition: Raises pH, ↑ unionized fraction, ↑ onset speed by 30-50%
- Epinephrine: Vasoconstriction prolongs duration 50-100%, ↓ systemic absorption
- Dose: Higher concentration → faster onset (more molecules available)
⭐ Clinical Pearl: Adding epinephrine 1:200,000 (5 μg/mL) to lidocaine extends duration from 1-2 hours to 2-4 hours and increases maximum safe dose from 4.5 mg/kg
Continue reading on OnCourse
Sign up for free to access the full lesson, plus unlimited questions, flashcards, AI-powered notes, and more.
CONTINUE READING — FREEor get the app
Have doubts about this lesson?
Ask Rezzy, our AI tutor, to explain anything you didn't understand
Practice Questions: Central Nervous System Pharmacology
Test your understanding with these related questions
Erenumab was approved by FDA in 2018 for which condition?